Dr.Sruthi, Dr.C.Jayakumar, Dr.Praveena
AIMS, Kochi
Two month old female 2nd born of a 3rd degree consanguineous parentage presented with poor weight gain since birth and feeding difficulty since 1 month
Birth weight of 2.6kg and born LSCS, due to oligohydramnios. Ba cried soon after birth. At birth, itself ba has generalised icthyosis. Ba had uneventful postnatal period
Weight dropped to 1.9 kg ba presumably due to inadequacy of breast milk and on started on formula feeds . Ba gained weight at 1 month (2.76kg).
At 1.5 months of life ban was admitted and parenteral antibiotics were given for cough.
At this point ba was advised to shift to Paladi feeds from bottle feeding
This was followed regurgitation ,diarrhoea poor weight gain and increased frequency of cough
Ba didn’t have social smile and had only received BCG vaccination. There is family history suggestive of elder sibling (4.5 year old male child) with developmental delay and dysmorphism proven to have congenital disorder of glycosylation type 1q.
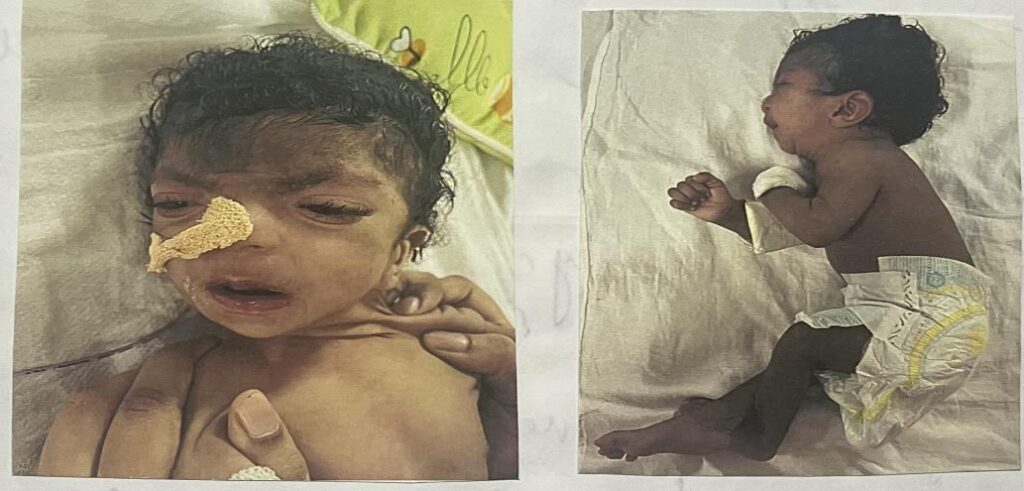
On examination ba appeared pale ,dysmorphic with severe washing and stunting but with stable vitals.Irrespective microcephaly anterior fronatanel was opened with short upturned nose, low set ears, hypertelorism ,long palpebral fissure , downturned mouth, macrostomia, long philtrum , thin upper lip, tongue tie, microretrognathia , camptodactyly of fingers , long toes and fingers, cutis marmorata over lower limb with loose skin folds over limb.
Ba was following of light with normal eye movements, bilateral wasting, variable tone with axial hypotonia, deep tendon reflexes – 2+ in all limbs. Other systems were within normal limits.
Labs CBC unremarkable
negative inflammatory markers
Chest X-Ray showed increased bronchovascular markings.
Peripheral smear reported as normocytic normochromic anemia with occasional reactive lymphocytes.
USG abdomen showed no sonological abnormalities.
ECHO done was normal.
MRI brain showed prominent supratentorial sulcal spaces and ventricles and hypoplastic inferior cerebellar vermis.
Eye evaluation showed bilateral flecked retina with severe optic disc hypoplasia which had a guarded visual prognosis. BERA, EEG and nerve conduction study were normal.
Swallow study revealed ba was unfit for oral feeds and hence nasogastric feeds were continued.
All these clinical features and dysmorphism seemed to fit in with congenital disorder of glycosylation 1q which the elder sibling has. Whole exome sequencing was thus done and it was reported to be consistent with the clinical suspicion of congenital disorder of glycosylation type 1q (SRD5A3 mutation on exon-1, homozygous, pathogenic for congenital disorder of glycosylation, type Iq).
Hence multidisciplinary approach was sought and kept under regular follow up with supportive management.
A genetically and clinically diverse collection of over 130 diseases known as congenital disorders of glycosylation (CDG)are brought on errors in different stages of the glycan modification pathways.
A great majority of these monogenic diseases exhibit several systemic symptoms, including variable coagulation, endocrine abnormalities, growth failure, developmental delay, and facial dysmorphisms. Their inheritance is mostly autosomal recessive.
The first-line screening test for patients with suspected CDG is serum carbohydrate deficient transferrin (CDT) analysis; However, its detection range is restricted to N-glycosylation abnormalities with deficiencies in sialic acid.
Genetic testing and dolichol-linked glycan analysis are examples of next-line testing. Since some of these rapidly expanding illnesses are curable, early detection is crucial.
Supportive therapy is the primary approach to treating glycosylation abnormalities, however targeted medicines are available for MPI-CDG, SLC35C1-CDG, PIGM-CDG, and PGM1-CDG.
The impact of consanguineous marriages on the prevalence of genetic diseases cannot be understated.
While such unions are culturally and historically significant in many societies, they also elevate the risk of inherited disorders due to the increased likelihood of shared genetic mutations.
This heightened risk underscores the importance of genetic counselling and screening for couples considering consanguineous marriage, as it enables informed decisions about family planning and reduces the burden of hereditary diseases on future generations.
By raising awareness and promoting informed choices, we can mitigate the adverse effects of consanguinity on public health and contribute to the well-being of individuals and communities worldwide.