
Dr. Terencia, Dr Sathyajith G Nair, Dr. SajithaNair, Dr. Sindhu, Dr. Sreya Nair DrC Jayakumar
Amrita Institute of Medical Sciences, Kochi
One year and six month old Non resident Indian child with history of recurrent respiratory infections since early infancy presented with wet cough, breathing difficulty and low grade intermittent fever of one dayduration.
Child is a late preterm 3rd child of a non consanguineous parentage (NCM)with a birth weight of 2.7kg. Around 4 hours of life child got ICU admitted for clinical and radiological pneumonia and 10ml blood was removed in After 5 days of NICU stay child was shifted to mother side and was discharged on day 18 of life on exclusive breast feeding.
Parents give history of recurrent respiratory infections through out infancy which was managed conservatively on OPD basis with nebulized bronchodilators and other supportive care.
Recent episode-Child was admitted in view of acute bronchiolitis and pneumomediastinum 2 weeks back and was treated with oxygen support, IV antibiotics, nebulized bronchodilators and other supportive care.
Child was evaluated for cystic fibrosis and Sweat Chloride done there on 09/03/2023 was negative.
Developmental History : Delay in gross motor milestones was present.
Gross motor- Stands with support(DQ-50%), Fine motor- Scribbles, Language-8-10 words, Social-Plays ball game
Vaccination History :Child is Vaccinated upto 1 year of age according to UAE schedule.
Family History- 3rd child of NCM with maternal asthma. No other bone or lung related disorders in other family members
Picture Widening of wrist
At admission This dysmorphic child was sick and pale looking, febrile, with respiratory distress.
Vitals:Febrile 100F, PR – 160/min, RR – 60/min, SPO2 – 90%in RA 97% in 4L O2(via O2 mask),CRT<3 sec
Pallor + Clubbing grade 2 + No (icterus, cyanosis, lymphadenopathy, edema).
Wide open anterior fontanelle(4*4cm),
Pre-auricular Ear tag in left side,
Depressed nasal bridge with bulbous nose tip,
Long eye lashes,
Prominent axillary field,
Prominent chest(Pectus carinatum) with rachitic rosary,
Wrist widening present.
Auxology Weight- 7.4 kg(b/w 0 and -1 SD) grade 1 PEM and grade 2 wasting, , Length- 75 cm(between 0 and -2 SD) grade 1 stunting,HC – 45.5 cm (b/w 0 and 1 SD),BMI (B/W -2 and -3SD), US:LS-1.1:1
RS : Tachypnea with subcostal and intercostal retractions present, Bilateral wheeze and scattered crepitations Iin all lung fields present.
P/A – Soft, non tender,no guarding,no organomegaly, BS+
CNS -Fully conscious,no cranial nerve deficit, mild hypotonia, Reflexes-brisk, No involuntary movements or meningeal signs of irritation.
CVS – No cardiomegaly,S1, S2 heard, no murmurs
Child was stabilized in the emergency and was started on oxygen support was started via Oxygen mask(98% on 4l O2).
Initial labs showed neutrophilic leucocytosis(TC-20K, N/L-75%/13%) and elevated inflammatory markers(CRP-27.05mg/L).
Chest xray showed Bilateral
increased bronchovascular
markings and BL patchy
consolidation.
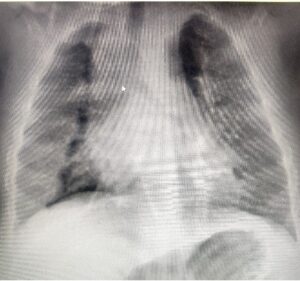
Fig 1-Chest Ray showing pneumonia
RFT,LFT, S.Electrolytes were within normal limits.
Child was shifted to PICU and was started on Inj Ceftriaxone, nebulized bronchodilators and other suppotives.
CT chest showed Segmental collapse/ patchy consolidation of posterior segment of right upper lobe, medial segment of right middle lobe and Bilateral lower lobes show subsegmental atelectasis.
Stool fat was positive ,As fecal elastase was low hence supplements started .
Vitamin D supplements was given in view of low vitamin D levels VITD
USG abdomen normal
Neurodevelopmental therapy was given
GE scintigraphy done showed no reflux. As per Genetics consultation X-rays-pelvis, forearm, foot and spine which showed Severe rachitic changes, small metaphysis with cupping, fraying and severe osteoporosis and bloods Calcium-9.6, Phos- 2.1, (PYLES INDEX calcium *phosphorous= 20.16 )ALP-636whichwas suggestive of hypophosphatemic rickets
WES sent and confirmed pathogenic mutation in SLC34A1 gene(Hypophosphatemic nephrolithiasis/osteoporosis).
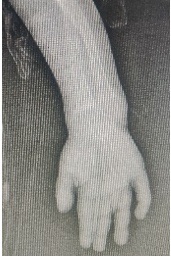
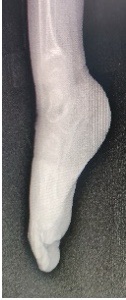
Fig 2 and 3- Xray extremities showing Severe rachitic changes, small metaphysis with cupping, fraying and severe osteoporosis
Endocrinology review was sought and chilsd was started on oral administration of phosphate and calcitriol along with vitamin D. Phosphate was administered in four to five doses per day, spaced at similar intervals through the waking hours(40 mg of elemental phosphorus/kg per day). Child was discharged on the same supplements and was kept in close follow up with endocrinology and pediatric gastroenterology.
Hereditary hypophosphatemic rickets refers to several inherited disorders characterized renal phosphate wasting, the most common of which is X-linked hypophosphatemia (XLH). This is a dominant disorder caused a variety of loss-of-function variants in the PHEX gene.
Feature is renal phosphate wasting, mediated excess fibroblast growth factor 23 (FGF23) activity.
XLH is characterized hypophosphatemia, slow growth, and rickets or osteomalacia; both males and females are affected
Disease onset is in childhood and presents with rickets and/or osteomalacia that is associated with hypophosphatemia, short stature, and secondary absorptive hypercalciuria.
An adult-onset form of the disease has been recognized in patients who were heterozygous carriers of SLC34A3 gene variants and presented with markedly reduced bone density, multiple fractures, hypophosphatemia, and hypercalciuria More commonly, heterozygotes manifest milder forms of HHRH, with mild hypophosphatemia, hypercalciuria, and nephrolithiasis but no signs of bone disease; however, this form may be underdiagnosedand is less well characterized.
Patients with HHRH should be treated with phosphate supplementation alone, using the same dosing schedule as described for X-linked hypophosphatemic rickets.
Endogenouscalcitriol levels are elevated, and the addition of exogenous calcitriol may be harmful.
Thus, plasma calcitriol levels and urinary calcium excretion should be measured before initiating therapy. Phosphate replacement therapy appears to normalize the serum phosphate concentration within a few days and improves but does not cure the osteomalacia.
Children should be seen every three months to monitor height; serum concentrations of calcium, phosphate, alkaline phosphatase, and creatinine; and urinary calcium excretion.
Renal ultrasonography should be performed once per year to evaluate nephrocalcinosis. A hand radiograph should be obtained once per year to exclude the reappearance of rickets and to determine bone age.
Patients with XLH can be treated with phosphate and calcitriol, which has been the standard treatment for XLH for decades.
The phosphate is given to replace renal losses, and calcitriol is necessary to increase the intestinal absorption of phosphate and calcium and to prevent secondary hyperparathyroidism. This therapy is continued at least through adolescence; it is controversial whether adults should continue therapy on a routine basis.