
Dr Rithwik Sunil, Dr Sheela Nampoothiri, Dr C Jayakumar
Three months and 3days old male 1st born of NCM brought with c/o of dysmorphism with antenatally detected left isomerism/intermediate AV canal defect (Large inlet VSD)
Child is feeding well, weight gain+ , no suck rest suck cycle,
No fast breathing, no chest indrawing noted, recurrent respiratory infections
Antenataly:
History of one abortion ,elderly primy with hypothyroidism (on medication) and antenatal diagnosed of VSD at 5month of gestation ,
Term/ CIAB/ LSCS(NPL and fetal bradycardia)/ Birth wt :2.8kg
Postnatal: after delivery admitted in NICU in view of cyanosis kept on O2 for 2days later started on feeding and discharged on DBF 5th DOL.
Immunization: appropriate for Age as per NIS schedule
Developmental: smiling +, head holding not attained, eye contact and recognizing mother. vision and hearing is normal . babble intermittently
sucking is good
no h/o of regurgitation
Examination
Vitals
PR; 132bpm
RR: 34cpm
Spo2 in lower limb: 96% on room air
NO Neurocutaneous markers
Head to foot examination:
AF open
Open mouth appearance
Triganular facies, high arched palate, wide nasal bridge
b/l dysplastic pinna
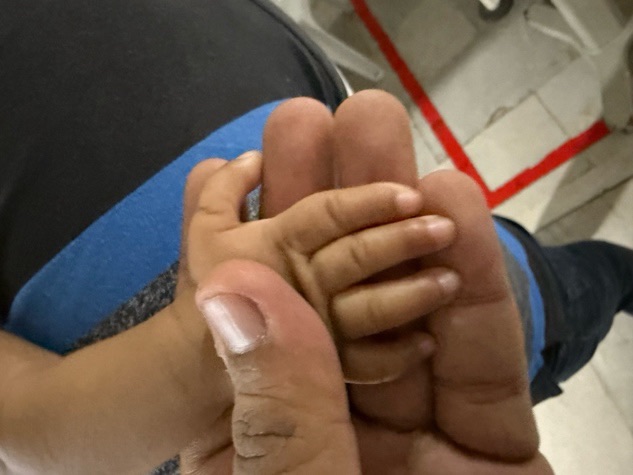
b/l preplaced thumb
right hypoplastic thumb
right UL short 2cm
there is ulnar deviation of right forearm
asymmetry of axillary folds
asymmetry of chest (rt elevated >left ) ? absent right pectoralis major
right axillary fold appears hypoplastic
LL normal
right testis undescended
a skin tag in the xiphisternal region

CVS :
S1 S2 heard, Grade 3 ESM +
RS :normal
Abdomen :
liver 2cm at sub coastal region
CNS :
Alert and active
Cranial Nerves : normal
Both pupils equally reactive to light, cough +,cry +
Hypotonia present (axial >appendicular)
head lag +,vertical suspension +,.
power -3/5 UL and LL reflexes :+1
planter Up going +
Auxology
Wt :4.13kg <1st Height :58cm( 3-50)
OFC 39cm (3-50th)
Investigation
ECHO
Restrictive inlet VSD; unrestrictive posterior muscular VSD (Left to right) Primum septum intact, two separate AV valves Moderate fossa ovalis ASD (Left to right)
XRAYs done
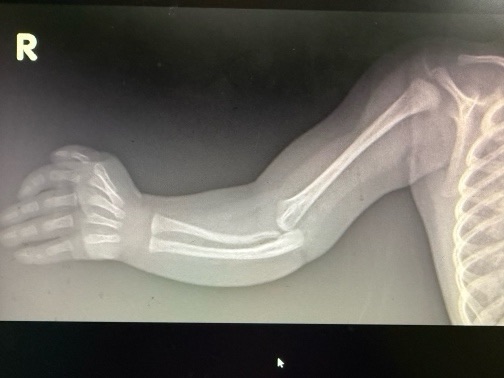
Showed right radius shortening
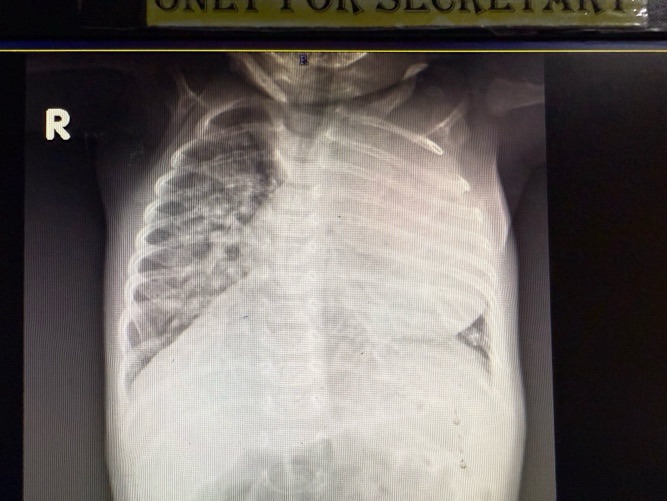
Cardiomegaly
Differential Diagnosis
Holt oram Syndrome
Fanconis Anemia
Thrombocytopenia absent radius syndrome
Teratogen exposure(thalidomide, valproate)
Duane Radial ray syndrome
Long thumb brachydactyly
Genetics Consult was availed and WES was sent
Child was seen Pediatric Cardiology Dept and started on Furoped, Digoxin and Aldactone
Corrective surgery is planned at the earliest available date
Holt Oram
Holt-Oram syndrome also referred to as the heart-hand syndrome, is an autosomal dominant disorder that is characterized upper limb abnormalities in association with congenital heart lesions.
A heterozygous mutation in the TBX5 gene on chromosome 12q24.1 causes Holt-Oram syndrome. This gene is responsible for encoding a transcription factor, T-box5, which regulates the expression of other genes in the development of the heart and limbs. Specifically, the gene is an important factor in cardiac septation and the development of bones in the arm and hand. More than 85% percent of individuals diagnosed with Holt-Oram syndrome carry the mutated TBX5 gene.
The condition is an autosomal dominant disorder, so one copy of the mutation in each cell will lead to the development of the syndrome. For an affected parent, this means that he or she has a 50% chance of transmitting the mutation to the offspring. However, most cases of Holt-Oram syndrome are sporadic and occur via de novo mutation.
There is complete penetrance for upper-limb abnormalities and 75% penetrance for cardiac defects. If the gene is inherited in an autosomal manner rather than de novo mutation, the offspring will often manifest greater severity in cardiac and limb deformities.
Life expectancy for Holt-Oram syndrome varies among affected individuals and predominantly depends on the severity of the congenital heart defect.