
Dr Joepaul Joy, Dr Vinayan KP,Dr Vyshak Anand(Ped neuro), Dr Sheela Nampoothiri(Genetics), Dr C Jayakumar AIMS, Kochi
Five year old male child, term, AGA, first of non consanguineous marriage with antenatal history of reduced fetal movements and fever in third trimester presented with high grade fever and wet cough since last 20 days. Child also had fast breathing and chest indrawing since 4 days.
Poor response to treatment was the train for referral to AIMS
Second child is 2 years old with history of status epilepticus and is on phenytoin . Postnatally child cried after suction and stimulation. Cry was weak and had poor activity. Urine and meconium passed within 24 hrs. No h/o suck rest suck cycle/ forehead sweating/ head nodding.
On day 24 of life during regular postnatal visit, child was noticed to have weight loss. Child started gaining weight after top feeds and around 40 days child became 3 kgs. Cry continued to be weak and poor sucking persisted. H/o easy slip through was present. No h/o regurgitation of feeds/ abnormal movements/ up rolling of eyes. Child was evaluated for cardiac problem and found to have patent foramen ovale and bicuspid aortic-valve.As time progressed mother noticed that there is delay in attaining age appropriate milestones. By 9 months child was evaluated for the same. MRI Brain done was normal and child was started on physiotherapy after which parents noticed significant improvement.
H/o difficulty in chewing and swallowing. GE Scintigraphy showed Grade 2 reflux. H/o febrile seizures at 1.5 years. There is history slight delay in all domains
On examination child was conscious and alert with stable vitals. Head to foot examination revealed Myopathic facies with small haed, long thin facies, low set large ears, left ptosis, high arched palate, overriding of toes, long 3rd toe, small little finger and hyperextensibility of joint present. Auxology is normal
Systemic examination revealed right sided crackles and wheeze with generalized hypotonia and absent reflexes.
Differentials
Congenital myotonic dystrophy,
Centronuclear myopathy,
Central core disease,
Multiminicore disease
CXR showed right paracardiac infiltrates. Child was started on oral azithromycin and IV Ceftriaxone given for 5 days.
Mantoux and TB GeneXpert done were negative.
Ophthalmology consultation availed and opined as mild ptosis not requiring active intervention.
Genetics opined for DNA isolation and further genetic test on review in OPD.
Xray Hip to rule out Ehler Danlos syndrome was normal. CPK was within normal limits. Child was discharged with stable vitals. WES done later showed autosomal recessive centronuclear myopathy, two variants in SPEG gene, one pathogenic and another VUS. Parental segregation report awaited. Parents were councelled in detail and advised to treat promptly for all respiratory infections, Pneumococcal and H Influenza vaccines and Physiotherapy.
I
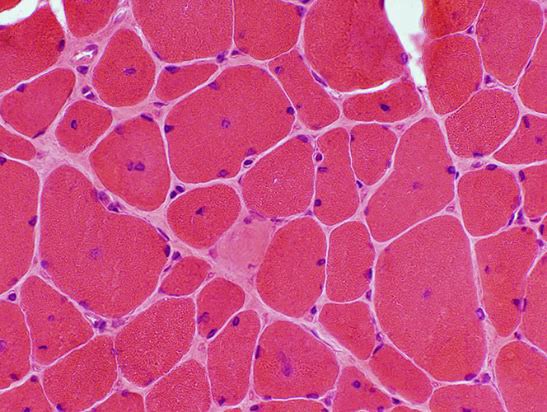
The centronuclear myopathies are a clinically and genetically heterogeneous group of disorders characterized muscle fibers with large central nuclei that resemble myotubes, the early fetal muscle fibers.There are three forms of inheritance for the centronuclear myopathies:
●Autosomal dominant ●Autosomal recessive ●X-linked myotubular myopathy
Centronuclear myopathies are genetically heterogeneous, with causative pathogenic variants described in the DNM2, MTM1, RYR1, BIN1, and TTN genes. The less common form occurs with autosomal dominant or recessive inheritance and consists of relatively mild weakness and hypotonia that may be unrecognized in the neonatal period. This form occurs in both males and females. The diagnosis of centronuclear myopathies is typically made genetic testing using a next generation sequencing targeted gene panel, whole exome sequencing, or whole genome sequencing. Single gene testing including deletion/duplication testing as well as genetic testing for familial pathogenic variants is available. A muscle biopsy may still be of value, particularly if variants of uncertain significance in the disease genes are identified. Creatine kinase usually is in the normal range, although occasionally it is elevated. Myopathic changes may be seen on the electromyography.