
Dr.Subbulakshmi P S, Dr.Sheela Nampoothiri, Dr.Dhanya Yasodharan, Dr.Jayakumar C
AIMS, Kochi
Eleven years old female child, presented with motor delay ,speech delay and recurrent respiratory infections from 6 months of age, Evaluation of LRTI at outside hospital revealed Cardiomegaly clinically radiologically .
She’s the third child of non consanguineous marriage.
Except for the weak cry since birth her perinatal history was normal
She was able to sit unsupported if made to sit and can elevate both hands above shoulder only at 2 years of age
Her mental development was normal
History of Sibling death ,who was similarly affected at 3.5 years is also told.
No work up was done for the ba at that time.

Growth parameters were less than third centile (failure to thrive+).
Immunisation’s were up-to date according to NIS.
Child was dysmorphic with open mouth appearance, bitemporal hallowing and a vague starring look.
The vital parameters were within normal limits.
Systemic examination showed no murmurs but cardiomegaly Generalised Hypotonia. Liver span measured was 8.5cm
DIFFERENTIALS:
● Glycogen storage disorder like Pompe’s disease, Forbes/Cori disease
● Congenital Muscular dystrophy like Duchenne
● Myocarditis
● Peroxisomal disorders
● Mitochondrial/Respiratory chain disorders
● Atonic CP
In view of cardiomegaly, frequent respiratory infections, hypotonia, developmental delay, Failure to thrive, boderline hepatomegaly and feeding difficulties Pompe’s was strongly suspected and she was investigated as follows.
Initial Labs:
SGOT/SGPT : 340/190.8
Creatinine Kinase: 1193
LDH: 1445
CRP: 4.6
TC: 11.6, N: 31, L: 48, Hb: 12.3, Platelets: 467
Enzyme analysis was done which showed deficiency of Alpha glucosidase consistent with atypical Pompe.
Echo showed Severe left ventricular hypertrophy (concentric hypertrophy) with EF of 68%.
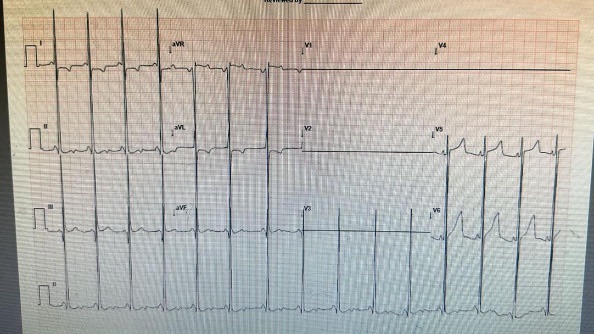
ECG: HR: 100/min, PR: 80ms, R/S in V1: 15/30 and in V5: 42/15(Short PR interval and broad and talk QRS complex)
The parents were counselled and advised to plan for Enzyme replacement therapy with alpha glucosidase which they agreed to receive once in 2 weeks and was being supported the charity trust. Enzyme Replacement Therapy (ERT) was started as 4 vials as infusion every 2 weeks.
Currently she’s 11 years old on regular ERT infusion and can stand with support with drastic clinical improvement.
DISCUSSION:
Pompe disease is Type 2GSD of glycogen storage disease, in which a complex sugar called glycogen builds up in your body’s cells.
The disease is due to deficiency of acid alpha-glucosidase (GAA).
GAA normally breaks down complex sugars
A lack of this enzyme causes glycogen to build up within the lysosomes.
This buildup occurs especially in heart and skeletal muscles
TYPES of Pompe :
INFANTILE ONSET: most severely affected.
A complete lack of, or very little, acid alpha-glucosidase enzyme causes this type of Pompe disease.
Symptoms start within the first year of life, most often around 4 months of age.
Symptoms and signs are severe muscle weakness, Hepatomegaly and sardonically due to cardiomyopathy.
The progression of the disease will happen quickly.
Without treatment, babies usually die of heart or respiratory failure between 1 and 2 years of age.
LATE ONSET(JUVENILE OR ADULT ONSET) : Late-onset Pompe disease can appear at any age — before 1 year of age but without cardinal(which distinguishes it from infantile-onset), but occurs later during adolescence or in adulthood.
Late-onset type have a reduced amount of acid alpha-glucosidase. This type is usually milder, and progression happens at a slower pace.
But it depends on the age of onset.
Children who develop symptoms are usually more severely affected. Symptoms include myopathy which lead to breathlessness and morality of due to respiratory failure
Pompe disease causes progressive muscle weakness, especially in the skeletal muscles like hips, legs, shoulders, arms and diaphragm.
Babies may have poor muscle tone
In addition,cardiomegaly,hepatomegaly and macroglossia
Other symptoms:
● Failure to thrive
● Respiratory infections
● Hearing problems
● Feeding issues
● Breathing difficulty
DIAGNOSIS:
Genetic testing for enzyme levels and measuring Creatinine kinase.
Echo, Electrocardiography, Electromyography, Pulmonary function tests can also supplement.
TREATMENT: Pompe disease treatment includes enzyme replacement therapy(ERT) with Algucosidase alfa or avalglucosidase alfa.
TAKE-HOME MESSAGE:
Without early detection and treatment of babies with infantile onset Pompe disease frequently die of respiratory or heart failure. People with late onset type of Pompe disease can often live longer as the disease progress at slower rate. However it depends on the age of onset and severity of the disease. Hence early detection and prompt treatment is necessary and the parents with a previously affected child must be given a proper prenatal counselling before the next conception.