
Dr Mahak, Dr Jayakumar, Department of Pediatrics, AIMS Kochi
Five month old male brought for abnormal head shape for evaluation.
Born at 38 weeks via LSCS due to Fetal Distress and meconium stained amniotic fluid. Head circumference 32.5cm kgs and3.2 kg birth weight
He was started on O2 hood at 2L for 4.5 hours and was noted to have 2 episodes of hypoglycemia on day 1 of life. He was thus maintained on OG Feeds for 1 day and shifted to mother’s side Day 2 of life.
As child was noted to have polydacytyly of right thumb with low set ears and he was evaluated further. USG Abdomen done was normal. NSG done showed mild disproportionate prominence of frontal horn of right lateral ventricle <10mm.
OAE done was normal. Tandem mass spectroscopy and chromatography were also normal.
He was discharged on breastfeeds and formula feeds Day 3 of life. Retinopathy screening done on follow up was also normal.
Ba was found to be growing well. Social smile was attained and 2 months with cooings 3 months.
At 3 months, he was evaluated further due to dysmorphic featured of high arched palate, large scaphoid head, low set ears, prominent scalp veins and right thumb polydactyly.
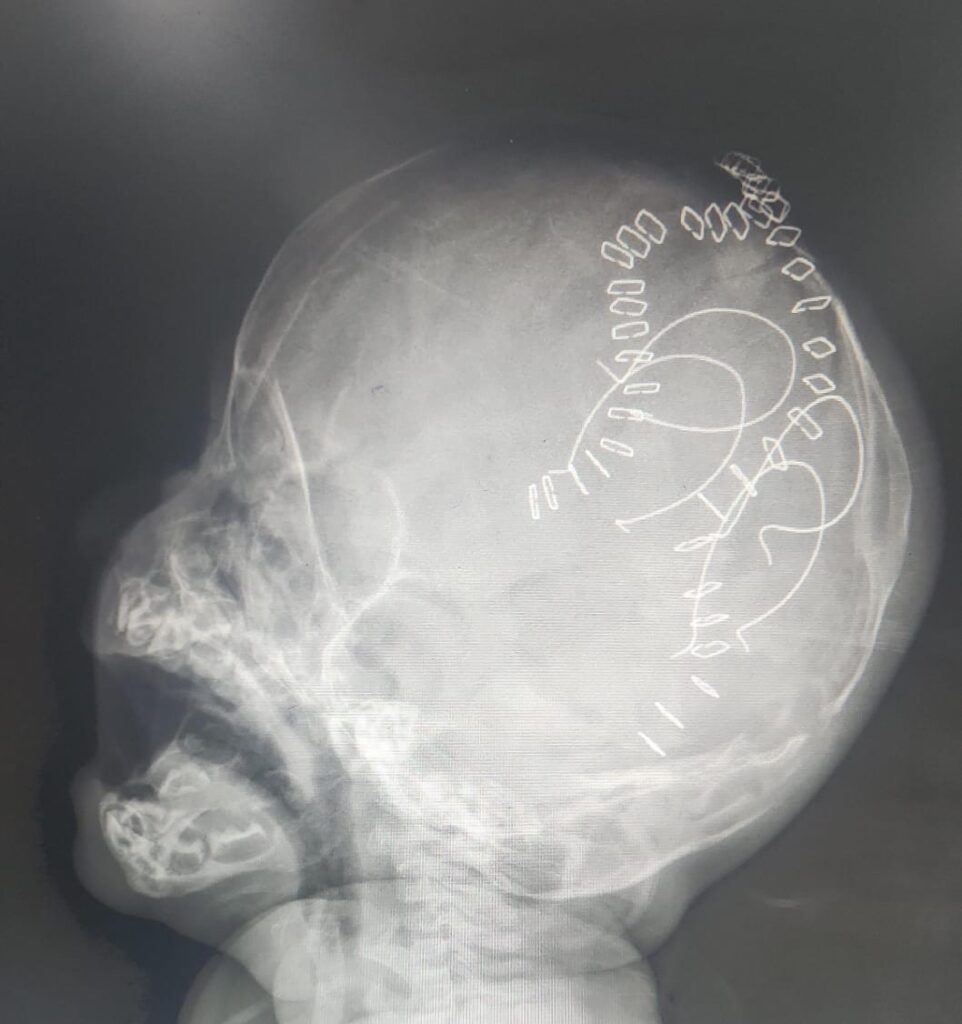
BERA done was normal. Xrays of whole body were normal. Echo showed a small OS ASD, Left to Right shunt. CT Brain done showed craniosynostoses with fused saggital sutures.
At presentation, child had a large head? Scaphocephalic in shape, Head circumference being 38cm, low set ears, Wide open AF, high arched palate, right hand thumb polydactyly, and a sacral dimple.
Differentials at this point–
Aperts Syndrome
Crouzon’s Syndrome
Carpenter’s Syndrome
Pfeiffer’s Syndrome
Saethze Chotzen Syndrome
Whole exome sequence was done which showed duplication of 5q34-q35,3 consistent with Carpenter’s syndrome. Child underwent Fronto Orbital Advancement and repair. Post operafive period was uneventful.
Craniosynostosis is premature fusion of one or more cranial sutures which causes growth parallel to the affected suture and not perpendicular to it.
It is seen in both syndromic ans non-syndromic children.
In syndromic, children, it is frequently associated with FGFR2 ans FGFR 3 mutations.

Carpenter’ Syndrome is described as Acrocephalopolysyndactyly Type 2 and is an autosomal recessive disorder associated with RAB23 mutation.
Clinical Features-
Brachycephaly and Scaphocephaly as bicoronial synostoses is associated with concurrent saggital/coronal/ lambdoid sutures.
Other features seen are hypoplastic mandible and/or maxilla with low set maldormed ears , supranumerary digits, clinodactyly campylodactyly, hypogondasisn,, omphalociele. Patients are moderarely affected mentally and early release of craniosynostis with fronto-orbital advancement is the treatment of choice.